ДИНАМИЧЕСКАЯ СТЕРЕОХИМИЯ
, раздел стереохимии, изучающий влияние пространств. строения молекул на скорости и направление р-ций, в к-рых они участвуют. Осн. характеристикой пространств. протекания р-ции является ее стереоселективность (или стереоспецифичность), определяемая стереоэлектронными требованиями, участием соседних групп и стерич. требованиями.
Если исходные соед. и (или) продукты р-ции могут существовать в двух или нескольких стереоизомерных формах, часто наблюдается преимуществ. образование одного из возможных продуктов. Такое предпочтение одного пути р-ции наз. стереоселективностью (СС). Она определяется как отношение разности кол-в образующихся изомеров А и Б к их сумме, СС = (А — Б)/(А + Б). Протекание р-ции исключительно по одному пространств. пути из нескольких возможных наз. стереоспецифичностью, т. е. стереоспецифичной является полностью стереоселективная реакция.
Р-ции стереоизомеров обычно протекают по неидентичным направлениям, приводящим к продуктам разного хим. или пространств. строения. Наиб. сложный случай - р-ции быстро взаимопревращающихся конформеров, к-рые подчиняются Кёртина
[Кертина] - Гаммета принципу.
При участии в р-ции двух центров образование и разрыв связей возможны с одной стороны к.-л. мол. фрагмента (супраповерхностно, с сохранением или обращением конфигурации обоих центров) или с разных сторон (антараповерхностно, с сохранением конфигурации одного реакц. центра и инверсией другого). Направление р-ции определяется наиб. энергетич. выгодностью и принципом наименьшего движения - миним. изменений положений атомов и исходной электронной конфигурации, включая сохранение орбитальной симметрии (см. Вудворда-Хофмана правила
).
Стереоэлектронные требования. Заключается в необходимости определенной пространств. ориентации орбиталей образующихся и разрывающихся в ходе р-ции связей. Одно из осн. требований расположение атомов, участвующих в элементарном акте р-ции, в одной плоскости (копланарность) или на одной прямой (коллинеарность). При этом обеспечивается благоприятствующее р-ции макс. или миним. перекрывание участвующих во взаимод. орбиталей. Эти общие правила проявляются в стереохимии р-ций разного типа.
При бимолекулярном нуклеоф. замещении у насыщенного атома С атака реагентом происходит со стороны грани тетраэдра, противоположной уходящей группе. Переходное состояние тригональная бипирамида с радикалами R, R, R:, находящимися в экваториальной плоскости; заместитель X и уходящая группа Y занимают апикальные положения (см. Конформационный анализ
):
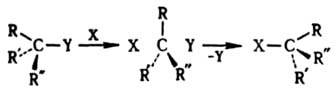
При этом рвущаяся и завязывающаяся связи коллинеарны. Происходит обращение конфигурации центр. атома [правило SN2-замещения Инголда (Ингольда)]. Обычно оно носит назв. вальденовского обращения по имени П. Вальдена, к-рый обнаружил в 1898 изменение знака оптич. вращения яблочной и хлорянтарной к-т, являющееся следствием обращения конфигурации в одной из последовательно проводимых р-ций:
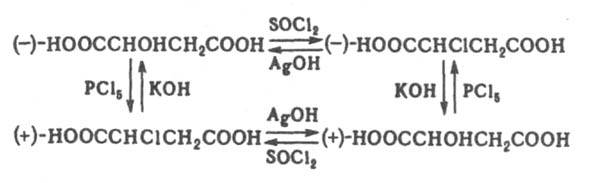
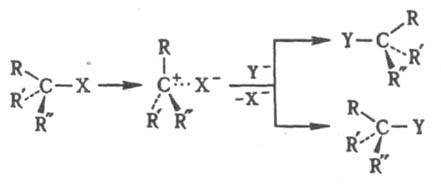
Мономолекулярное нуклеоф. замещение проходит через плоский интермедиат - карбкатион RRR:C+. Обычно возможна нестереоселективная атака с обеих сторон с рацемизацией (правило SN1-замещения Инголда). Если же в образовавшейся ионной паре уходящая группа затрудняет доступ к катионному центру со стороны разорвавшейся связи, наблюдается частичная инверсия конфигурации, причем степень рацемизации зависит от времени жизни карбкатиона в данных условиях:
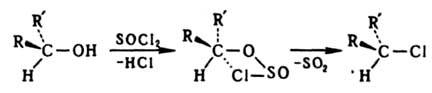
Сохранение конфигурации возможно при связывании реакц. центра со стороны, противоположной уходящей группе. При замещении по внутримолекулярному механизму SNi конфигурация сохраняется вследствие образования циклич. интермедиата, напр.:
Бимолекулярное электроф. замещение у насыщенного атома С идет через пентакоординированные интермедиаты разл. структуры. В случае ртутьорг. соед. конфигурация сохраняется, но в ряду кремний- и оловоорг. соед. наблюдается частичная инверсия конфигурации. Для мономолекулярного электроф. и радикального замещения характерна рацемизация. Электроф- и радикальное замещение у алкенового атома С протекает с сохранением конфигурации двойной связи (Несмеянова-Борисова правило), вероятно, через циклич. переходное состояние. Напр., изотопный обмен атома Hg идет по схеме (звездочкой обозначен меченый атом):
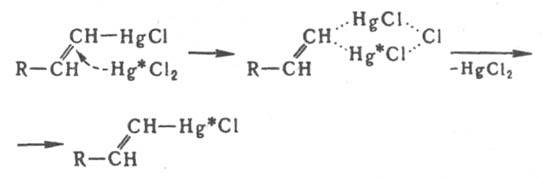
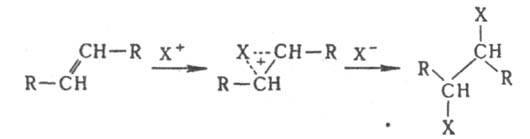
Присоединение электрофилов к кратным связям протекает антараповерхностно с промежут. образованием p-комплекса, в к-ром доступ к атомам С открыт только со стороны, противоположной направлению первоначальной атаки, так что образуются продукты транс
-присоединения:
Также реагирует тройная связь в р-циях с Вr2, НВr или при восстановлении. Каталитич. восстановление цис
-стереоселективно:
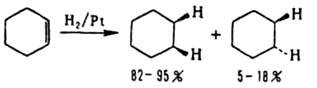
Супраповерхностное цис
-присоединение происходит при гидроксилировании олефинов под действием перманганата, при гидроборировании; оно включает циклич. интермсдиат или имеет многоступенчатый механизм.
При 1,2-элиминировании действует правило копланарности четырех центров: в бимолекулярной р-ции отщепляются группы, расположенные антипсрипланарно, что для циклич. соед. соответствует диаксиальной конформации (правило Е2-отщепления). Поэтому для разных конформеров характерны разл. р-ции элиминирования, напр.:
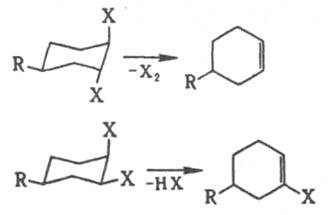
При неблагоприятном расположении отщепляющихся фрагментов молекула должна предварительно претерпеть конформац. переход. Элиминирование в этиленовом ряду с образованием тройной связи подчиняется тому же правилу. Мономолекулярное элиминирование идет через карбкатион, и стереохим. направленность р-ции зависит от его времени жизни. цис
-Отщепление через циклич. переходное состояние осуществляется, напр., при расщеплении ацетатов, ксантогенатов, аминоксидов.
Стереоэлектронные требования, в первую очередь сохранение орбитальной симметрии, играют определяющую роль в пространств. направленности перициклич. р-ций. Циклоприсоединение и обратная р-ция циклоэлиминирования могут протекать супра- или антараповерхностно (соотв. цис-
и транс
-присоединение). Диеновый синтез за редкими исключениями стереоспецифичен и дает продукты циc
-присоединения.
Участие соседних групп. Проявляется, напр., в содействии сохранению конфигурации в р-ции нуклеоф. замещения при наличии в субстрате групп В нуклеоф. характера (OR, SR, NR2, O-, OCOR, Hal, Ph), фиксирующих карбкатионный центр и находящихся в вицинальном положении к нему. Эффект этих групп заключается в образовании внутр. комплекса, так что в переходном состоянии экранирована та сторона карбкатионного центра, которая противоположна направлению движения уходящей (замещаемой) группы, и для атаки реагентом открыта только одна сторона:
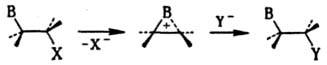
Фактически на первой стадии происходит "внутренняя" р-ция SN2, и в целом процесс включает две взаимокомпенсирующиеся инверсии конфигурации. При этом может резко увеличиваться скорость р-ции; напр., гидролиз b-хлорэтилсульфидов RSCH2CH2Cl идет в 104 раз быстрее гидролиза
аналогичных хлорэтиловых эфиров благодаря образованию эписульфониевого иона. Такое участие носит назв. анхимерного эффекта (анхимерного содействия), или синартстич. ускорения.
Стерические (пространственные) требования. Заключаются в необходимости определенного своб. пространства для подхода реагента, перемещения фрагментов молекулы и удаления уходящей группы при образовании переходного состояния или интермедиата. Стерич. препятствия, создаваемые группами, расположенными вблизи реакц. центров, зависят только от объема, но не от электронной природы групп и количественно описываются их стерич. константами (см. Корреляционные соотношения
). Стерич. требования влияют на образование переходного состояния или интермедиата, необходимого с точки зрения стереоэлектронных требований. Если в нем энергия стерич. препятствия выше, чем в исходном состоянии, наблюдается замедление р-ции, и наоборот. Напр., при мономолекулярном замещении у насыщенного атома С радикалы в плоском интермедиате RRR:C+ более удалены друг от друга, чем в тетраэдрич. исходном и конечном соединениях. Поэтому возрастание их объема в большей мере увеличивает пространств. затруднения в исходном состоянии, чем в карбкатионе, что приводит к понижению энергии активации р-ции. Напротив, в пентакоординированном переходном состоянии р-ции SN2 имеет место более сильное стерич. взаимод., чем в тетраэдрич. молекулах, и чем больше стерич. требования радикалов, тем выше относит. энергия переходного состояния, тем труднее оно образуется и тем медленнее протекает р-ция.
Кроме того, стерич. требования проявляются в экранировании реакц. центра, т. е. в создании пространств. препятствий для подхода к нему реагента. Напр., бициклич. соед. с заместителями в голове моста, такие как бициклооктаны (ф-ла I) и адамантаны (II), не вступают в р-ции типа
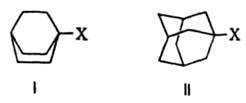
SN2 вследствие невозможности подхода нуклеофила к узловому атому С с "тыльной" стороны, закрытой бициклич. системой. Стерич. требования определяют и затрудненность мономолекулярной р-ции, т. к. интермедиат с плоским карбкатионным центром в голове моста чрезвычайно энергетически невыгоден.
При наличии нескольких стереоизомерных путей р-ции стереоселективность может возникнуть в результате несимметричного окружения двух- или многоцентрового реагирующего фрагмента, если к.-л. группы создают стсрич. препятствия для подхода реагента в одном из направлений. Напр., в переходном состоянии диенового синтеза возможна эндо-
или экзо
-ориентация заместителя X в несимметричном диенофиле по отношению к диеновой системе:
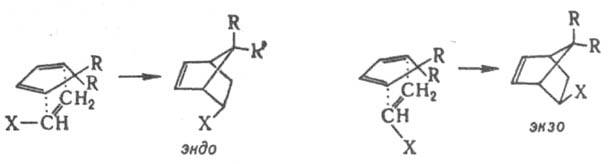
Стереоселективность этих и аналогичных р-ций определяется как электронными взаимод. орбиталей диена и заместителя X, так и различием стерич. условий в двух переходных состояниях. При R = Сl экзо
-ориентация невозможна из-за сильного сближения групп R и X. Стерич. препятствия обусловливают атаку двойной связи в бициклоалкенах с более доступной стороны, напр., в диеновом синтезе, эпоксидировании, присоединении карбенов и др.:
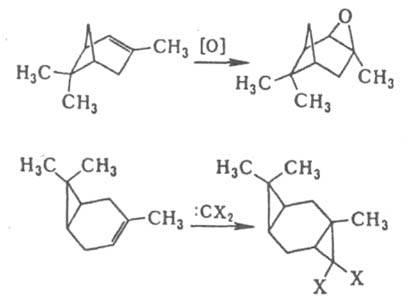
В производных циклогексанола экранирование цис-
аксиальными атомами Н приводит к тому, что аксиальный гидроксил ацилируется медленнее экваториального. По этой же причине затруднены р-ции SN2 при экваториальной уходящей группе.
Присоединение со стороны заместителя (син
-присоединение), наблюдающееся для ряда циклоалкенов с полярными группами, б. ч. обусловлено образованием промежут. комплексов, в частности, в р-циях эпоксидирования:
Изучение Д. с. необходимо при исследовании механизмов мн. хим. р-ций, а также позволяет находить оптимальные пути стереоселективиого синтеза. См. также Рацематы
.
Лит.: Соколов В.
И., Введение
в теоретическую стереохимию, М., 1979; Ногради М., Стереохимия, пер. с англ., М., 1984; Потапов В. М., Стереохимия, 2 изд., М., 1988. А. Н. Верещагин.
|


